 国际站
国际站 近日,北京建筑大学联合北京大学、华东理工大学在国际顶级期刊《Nature》子刊《Nature communications》(IF:16.6,中国科学院一区,TOP期刊,H指数248)上以长文形式发表题为“Multi-channel electron transfer induced by polyvanadate in metal-organic framework for boosted peroxymonosulfate activation”的高水平研究论文。

这是北京建筑大学首次以第一单位在该期刊发表论文,北京建筑大学环境与能源工程学院硕士研究生兰明岩和北京大学深圳研究生院博士研究生李渝航为共同第一作者,北京建筑大学环境与能源工程学院王崇臣教授、北京大学深圳研究生院冀豪栋助理教授和华东理工大学邢明阳教授为共同通讯作者。
该研究报道了一种多酸MOF(Co2(V4O12)(bpy)2)作为类芬顿反应催化剂,通过非自由基途径实现对具有高HOMO的富电子微污染物的高效降解。通过各种实验、表征和DFT计算,验证了在催化剂的界面处PMS与催化剂可形成多重主客体间的相互作用。证实了此活化机制也适用于其他POM-MOFs/PMS体系。这项研究为制备高效的含多酸催化剂用于水净化提供了研究基础,也为后续探究不同POM-MOFs用于类芬顿反应,并挖掘其在大规模水环境修复中应用提供了新思路。
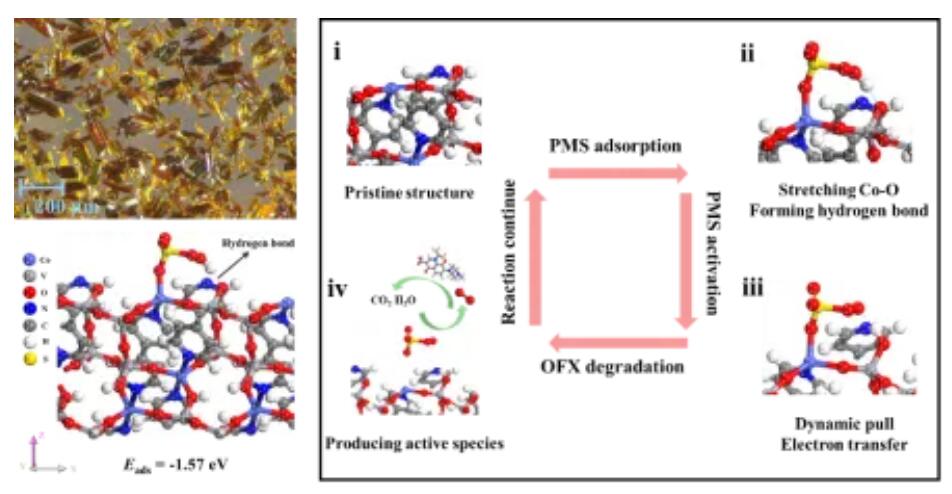
北京建筑大学环境与能源工程学院、环境修复功能材料北京市重点实验室主任王崇臣教授课题组,采用多金属酸簇([V4O12]4−)和4,4-联吡啶分别作为无机和有机配体,制备出一种钴基金属有机框架材料(Co2(V4O12)(bpy)2),与报道过的催化剂进行性能对比发现,Co2(V4O12)(bpy)2对具有高HOMO的富电子微污染物的催化降解效率甚至超过了许多已报道的单原子催化剂。此外Co2(V4O12)(bpy)2/PMS体系还具有良好的脱毒效果及实际应用潜力。通过构建不同PMS吸附模型进行几何结构优化得出最优吸附模型,从而具体研究PMS与Co2(V4O12)(bpy)2表界面的相互作用。结果表明部分电子从Co位点传递到PMS进行活化反应,另一部分电子被“电子海绵”[V4O12]4−捕获进而通过氢键通道传递向PMS。PMS的活化过程中的O-H键由于得到电子而发生断裂,导致PMS中的H与[V4O12]4−中的端基O之间形成了新的O-H键,生成SO5*进而生成1O2降解污染物。
该项研究由北京建筑大学环境与能源工程学院王崇臣课题组与北京大学深圳研究生院冀豪栋助理教授团队、华东理工大学邢明阳教授团队合作完成,北京建筑大学环境与能源工程学院为第一完成单位。北京建筑大学环境与能源工程学院王崇臣教授、北京大学深圳研究生院冀豪栋助理教授和华东理工大学邢明阳教授为共同通讯作者。北京建筑大学环境与能源工程学院硕士研究生兰明岩和北京大学深圳研究生院博士研究生李渝航为共同第一作者。中国科学院高能物理研究所马宇辉教授、北京建筑大学环境与能源工程学院硕士研究生李欣洁、孟令辉,华东理工大学曹嘉真和北京大学深圳研究生院高帅为共同作者。该研究得到了国家自然科学基金项目、北京建筑大学"头雁"计划及深圳市科技计划的支持与资助。

全文速览
在过渡金属(如Fe、Co、Mn)基催化剂的类芬顿反应体系中,一般认为过渡金属是激发氧化剂产生活性物种的主要电子供体。事实上,在众多复杂的催化剂的表界面处,过渡金属可能不是唯一的电子供体。因此,探究在类芬顿反应中氧化剂的活化过程有利于为后续设计催化剂和研究构效关系具有重要意义。本研究采用多金属酸簇([V4O12]4−)和4,4-联吡啶分别作为无机和有机配体,制备出一种钴基金属有机框架材料(Co2(V4O12)(bpy)2)。研究表明该催化剂通过活化PMS可产生非自由基单线态氧(1O2)和部分表面结合态硫酸根自由基(SO4•−),进而实现了对具有较高HOMO的富电子微污染物的降解。X射线吸收光谱和DFT计算发现催化剂中的Co位点作为PMS的吸附位点,但并不是主要的电子供体。通过一系列原位表征和DFT计算证实在催化剂表界面处,[V4O12]4−中的端基氧可与PMS的端基氢相互作用形成氢键。由于[V4O12]4−具有“电子海绵”效应,在“电子海绵”中储存的电子通过氢键定向传导到PMS的O-H键上实现SO5•−中间体的生成,进一步可生成1O2和SO4•−。这项研究不仅提出了一种高效的电子海绵催化剂可通过非自由基途径实现水净化,还为类芬顿反应的机理研究提供了新的见解。
图文导读
01、结构表征
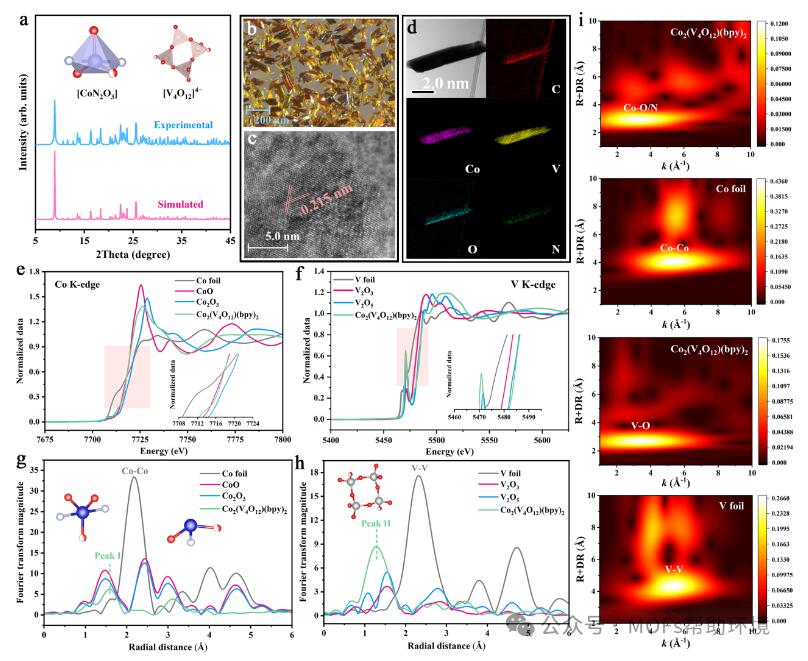
图1 Co2(V4O12)(bpy)2的(a)PXRD图;(b)光学显微镜图像;(c)HR-TEM图像;(d)TEM图以及相应的元素映射分布图。Co2(V4O12)(bpy)2的(e)Co K-edge XANES光谱;(f)V K-edge XANES光谱;(g)Co K-edge FT-EXAFS光谱;(h)V K-edge FT-EXAFS光谱图和(i)Co与V的小波变换图。
结合粉末XRD(图1a)和单晶XRD解析得出Co2(V4O12)(bpy)2的原子结构,其是由Co2+作为中心金属离子,桥联无机连接剂[V4O12]4−簇和有机连接剂4,4'-联吡啶(bpy)组成三维框架结构,五配位的Co2+桥联两个bpy的N原子和三个[V4O12]4−簇的O原子,每个[V4O12]4−簇中存在两个不配位的末端O原子。经光学显微镜观察,发现Co2(V4O12)(bpy)2呈粒径约200 μm的黄褐色晶体(图1b)。HR-TEM显示Co2(V4O12)(bpy)2的晶格尺寸为0.215 nm,为Co2(V4O12)(bpy)2的(3 3 2)晶面(图1c-d)。采用X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)分析得出Co2(V4O12)(bpy)2催化剂中Co和V的局部配位环境及电子结构性质。图1e-1f表明Co的价态接近+2,V的价态接近+5。由图1g-1h中Co和V K edge FT-EXAFS光谱可知,Co2(V4O12)(bpy)2中的Co原子与O原子和N原子配位。此外,对样品中的Co和V元素进行小波变换(WT)表征的结果表明,样品中不存在Co-Co键或V-V键(图1i),说明Co2(V4O12)(bpy)2的结构中不存在Co团簇。
02、性能研究
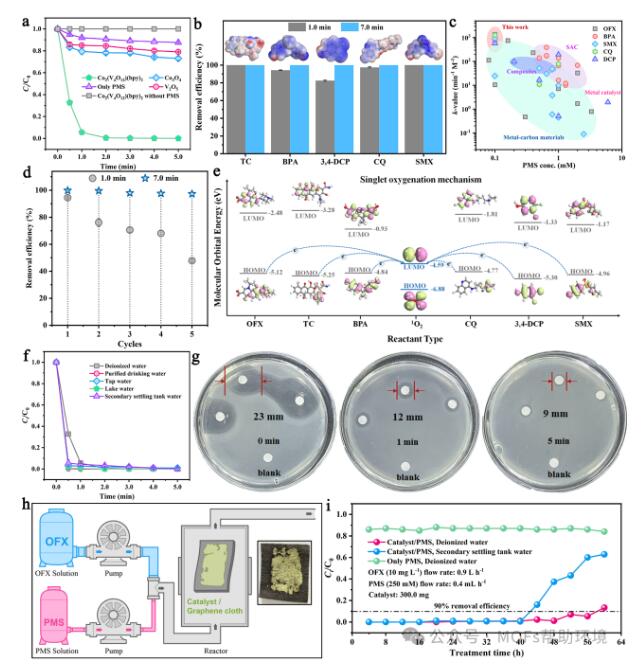
图2(a)不同催化剂的类芬顿反应活性测试。(b)Co2(V4O12)(bpy)2/PMS体系对TC、BPA、3,4-DCP、CQ和SMX的降解效率。(c)Co2(V4O12)(bpy)2与已报道过的催化剂的性能对比;(d)Co2(V4O12)(bpy)2催化降解OFX的循环实验。(e)1O2对多种富电子微污染物的氧化机理。(f)Co2(V4O12)(bpy)2在真实水体中对OFX降解效率的影响。(g)经Co2(V4O12)(bpy)2/PMS体系降解后的OFX溶液对大肠杆菌的抑菌圈图像。(h)连续降解OFX的固定床反应器(插图:Co2(V4O12)(bpy)2粉体固定在石墨毡的实物图像)。(i)不同体系在反应器中对OFX的降解效率。
选取富电子污染物氧氟沙星(OFX)作为目标污染物。实验结果(图2a)表明Co2(V4O12)(bpy)2具有优异的PMS活化效率,其降解效率远高于商品化的Co3O4和V2O5。此外,对于其它富电子污染物如四环素(TC)、双酚A(BPA)、3,4-二氯苯酚(3,4-DCP)、磺胺甲恶唑(SMX)和磷酸氯喹(CQ),Co2(V4O12)(bpy)2/PMS体系依旧可在7.0 min内实现对污染物的完全降解(图2b)。通过与报道过的催化剂进行性能对比(图2c)发现,Co2(V4O12)(bpy)2具有史上最快的催化降解效率,其催化降解效率甚至超过了许多已报道的单原子催化剂。循环实验(图2d)证实Co2(V4O12)(bpy)2具有良好的水稳定性和循环利用性。通过一系列捕捉实验、ESR定量、溶剂(D2O)替换实验、分子探针实验等证实Co2(V4O12)(bpy)2/PMS体系以1O2和部分SO4•−为主要的活性物种,1O2氧化OFX的贡献率约为84%。众所周知,富电子污染物可在非自由基系统中被选择性氧化。通过计算上述富电子有机污染物、苯甲酸(BA)和硝基苯甲酸(NBA)的最高占据分子轨道、最低未占据分子轨道和静电势发现,TC、BPA、3,4-DCP、CQ和SMX均具有明显的富电子基团且最高占据分子轨道全部高于BA和NBA(图2e),这有利于1O2进行提电子反应。由于非自由基氧化具有良好的环境抗干扰能力,Co2(V4O12)(bpy)2/PMS体系对饮用水、湖水、自来水和二沉池出水配置的模拟真实水体中的OFX同样可实现快速且100%的降解(图2f)。大肠杆菌(E. coli)抑菌性实验(图2g)表明,经催化氧化后的溶液的毒性远小于OFX溶液,证实Co2(V4O12)(bpy)2/PMS体系具有良好的脱毒效果。此外,我们将粉体催化剂固定在石墨毡上,构筑了连续流动装置(图2h)。使用纯水和二沉池出水配置含OFX的废水,在长达56 h和40 h的连续处理中,Co2(V4O12)(bpy)2对OFX的去除效率保持在90%以上,表明Co2(V4O12)(bpy)2具有良好的实际应用潜力(图2i)。
03、机理解释
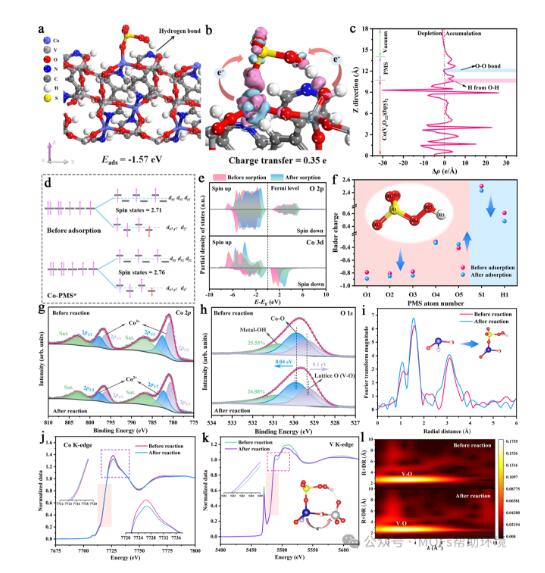
图3 (a)最佳PMS吸附模型及相应的吸附能。(b)最佳PMS吸附模型的差分电荷密度。(c)基于差分电荷密度的平面平均电子密度差Δρ (Z)。(d)PMS吸附前后Co 3d轨道的电子填充情况和自旋态变化。(e)PMS吸附前后O 2p 和Co 3d 的PDOS谱图。(f)PMS在吸附前后的Mulliken电荷变化。Co2(V4O12)(bpy)2在反应前后的(g)Co 2p和(h)O 1s的XPS谱图。反应前后Co2(V4O12)(bpy)2的(i)Co K-edge EXAFS谱图。(j)Co2(V4O12)(bpy)2的Co K-edge EXAFS谱图;(k)V K-edge XANES谱图和(l)WT图。
通过构建不同PMS吸附模型进行几何结构优化得出图3a为最优吸附模型,PMS中S连接的O原子与Co位点配位,末端H与[V4O12]4−的端基氧通过氢键相连。在最优吸附模型的基础上进行差分电荷密度计算(图3b),催化剂向PMS转移的电子数为0.35 e。平面电子密度差(图3c)显示电子的聚集区集中在氢原子附近而不是过O-O键。根据洪特定则和泡利不相容原理对Co位点的最外层d轨道进行解析(图3d),结果表明PMS吸附前后,自旋态并未发生明显变化,意味着Co位点并不是活化PMS的主要电子传输中心。而分析吸附PMS前后Co 3d、V 3d、O 2p的PDOS发现其电子结构性质均发生改变(图3e)。Mulliken电荷结果(图3f)表明,吸附后PMS模型中的H相比原始PMS模型获得了更多的电子,结合吉布斯自由能计算发现,PMS的活化过程倾向断O-H键生成SO5*。XPS分析结果表明,催化反应后Co3+的百分比从27.7%增加到33.6%(图3g),Co-N和Co-O键的结合能向更高能级移动(图3h),表明部分电子从Co位点传递到PMS进行活化反应,另一部分电子被“电子海绵”[V4O12]4−捕获进而通过氢键通道传递向PMS。Co K边EXAFS光谱显示PMS活化反应后Co-O键的强度和平均键长发生变化(图3i),这可能是由于Co-OPMS键的形成。反应后Co的白线峰强度变弱表示Co位点的部分电子被转移(图3j)。而反应后V金属的白线峰强度增强,表明V作为电子受体吸引电子导致电荷密度增加(图3k)。结合反应前后的WT图(图3l),表明Co-N/O键和V-O键在PMS活化反应过程中呈现动态变化,进一步验证前面提出的电子转移过程。
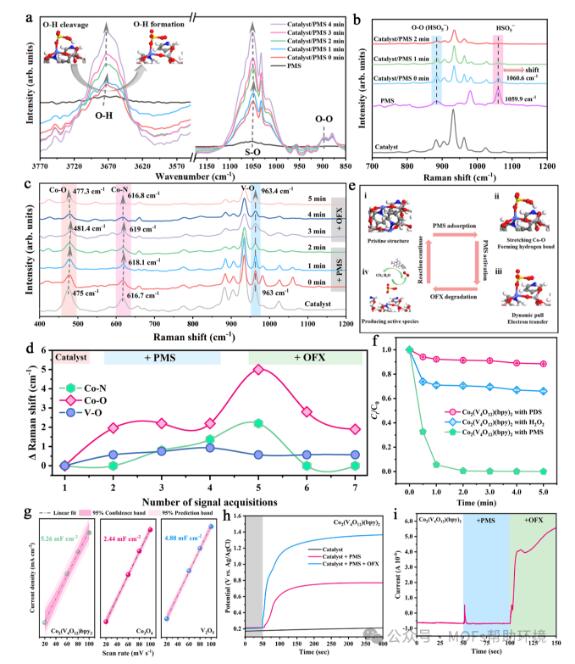
图4 (a)Co2(V4O12)(bpy)2/PMS体系在N2氛围下的原位红外光谱。(b)Co2(V4O12)(bpy)2、PMS及二者反应过程中的原位拉曼图谱。(c)原位拉曼光谱中不同金属键的伸缩变化。(d)不同金属键在反应中的伸缩振动位移。(e)类芬顿反应过程中PMS的活化机理及OFX氧化机理。(f)不同的活化剂对OFX降解效率的影响。(g)根据Co2(V4O12)(bpy)2、Co3O4和V2O5的CV曲线进行的ECSA测量。使用Co2(V4O12)(bpy)2作为工作电极的(h)开路电压曲线和(i)i-t曲线。
为了进一步研究在PMS活化过程中PMS与Co2(V4O12)(bpy)2表界面的相互作用,在N2氛围下进行了原位红外和原位拉曼测试。原位红外(图4a)结果表明,当PMS溶液中加入催化剂时,属于O-H键的峰迅速从3676 cm-1蓝移到3673 cm-1,随着时间的推移,峰值又逐渐趋近于3676 cm-1。O-H键的动态变化可归因于以下原因:(1)PMS中的O-H键与催化剂表面产生了强烈的相互作用;(2)随着反应的进行,PMS中的O-H键由于得到电子而发生断裂,导致PMS中的H与[V4O12]4−中的端基O之间形成了新的O-H键。这一结果也与XPS分析结果完全吻合,即类芬顿反应后Co2(V4O12)(bpy)2上的羟基含量明显增加。S-O键的峰发生轻微移动则可能是由于Co位点对PMS的吸附作用。在无溶解氧的水溶液和N2氛围下,在~896 cm-1处的O-O键出现了特征性伸展,这可证实Co2(V4O12)(bpy)2/PMS体系中生成了大量的1O2。原位拉曼谱图显示HSO5−的特征振动峰发生微弱的红移(图4b),这可归因于电子从Co2(V4O12)(bpy)2催化剂转移到PMS上。此外,通过原位拉曼光谱还观察到了催化剂中金属键的动态变化。光谱结果显示,与Co-O(476.8 cm-1)和Co-N(962.3 cm-1)键明显的动态变化相比,由于[V4O12]4−具有稳定的结构特征,V-O(619 cm-1)键的轻微动态变化可以忽略不计(图4c-d)。具体活化过程如图4e展示。除探究PMS的活化机制外,还选择了PDS和H2O2作为氧化剂进行类芬顿反应。基于本项研究结果,H2O2的末端H也可与[V4O12]4−簇的末端O产生相互作用,通过定向电子传递实现H2O2的高效活化;而PDS则不具备形成氢键的条件只能通过Co位点的电子传递活化PDS。正如预期,Co2(V4O12)(bpy)2/H2O2体系对OFX的降解效率强于Co2(V4O12)(bpy)2/PDS体系(图4f)。电化学表征也证实了Co2(V4O12)(bpy)2/PMS体系对于降解富电子污染物的优越性(图4g-4i)。
小结
本研究首次使用多酸MOF(Co2(V4O12)(bpy)2)作为类芬顿反应催化剂,通过非自由基途径实现对具有高HOMO的富电子微污染物的高效降解。通过各种实验、表征和DFT计算,验证了在催化剂的界面处PMS与催化剂可形成多重主客体间的相互作用。由于[V4O12]4−在Co2(V4O12)(bpy)2中的配位模式,使得[V4O12]4−的端基O与PMS的末端H之间形成氢键。同时,[V4O12]4−具有“电子海绵”的特性,可通过形成的氢键作为定向电子转移通道将电子转移至PMS。生成的SO5•−自由基进一步分解为1O2和SO4•−并用于降解去除微污染物。经验证,此活化机制也适用于其他POM-MOFs/PMS体系。这项研究为制备高效的含多酸催化剂用于水净化提供了研究基础,也为后续探究不同POM-MOFs用于类芬顿反应,并挖掘其在大规模水环境修复中应用提供了新思路。

王崇臣,男,博士,北京建筑大学教授、博士生导师。建筑结构与环境修复功能材料北京市重点实验室主任,Chinese Chemical Letters、Environmental Functional Materials等12个期刊副主编、编委。中国材料研究学会理事/副秘书长、北京化学会理事/副秘书长/科普委员会主任、北京环境科学学会理事/科技创新分会常务副主委、中国环境科学学会水处理与回用专业委员会委员、中国感光学会光催化委员会委员。入选北京市百千万人才、北京市高创计划百千万领军人才和长城学者。获得北京市高等学校青年教学名师奖。主要研究领域为环境功能材料、水文化。主持国家自然科学基金面上项目、北京自然科学基金重点(B类)/面上项目、北京社科基金重点项目等纵向项目10余项。入选全球高被引学者和中国高被引学者。


来源:北京建筑大学 ,爱科会易仅用于学术交流