 国际站
国际站 2024年8月23日,Science在线发表了帝国理工学院David Pfau课题组的研究论文,题目为《Accurate computation of quantum excited states with neural networks》。

理解物质如何与光相互作用的物理需要对量子体系的电子激发态进行精确建模。这是光催化剂、荧光染料、量子点、发光二极管(LED)、激光器、太阳能电池等的基础。现有的激发态量子化学方法可能比基态量子化学方法更不准确,有时在定性上也是如此,或者可能需要针对特定状态的先验知识。神经网络与变分蒙特卡罗(VMC)相结合,在一系列体系的基态波函数方面取得了显著的精度,包括自旋模型、分子和凝聚态体系。尽管VMC已被用于研究激发态,但现有的方法存在局限性,使其难以或不可能与神经网络一起使用,并且通常有许多自由参数需要调整才能获得良好的结果。
在此研究中,作者提出了一种通过VMC估计量子体系激发态的算法,该算法不需要自由参数,也不需要状态的正交化,而是将问题转化为寻找扩展体系的基态。可以计算任意的观测值,包括非对角线期望值,如跃迁偶极矩。该方法特别适用于神经网络方法,并且通过将该方法与FermiNet和Psiformer方法相结合,可以准确获得一系列分子的激发能和振子强度。在苯级分子上实现了精确的垂直激发能,包括具有挑战性的双激发。
自然激发态VMC(NES-VMC)是一个无参数且数学上合理的激发态变分原理。将其与神经网络方法相结合,可以在各种基准问题上实现显著的准确性。对量子系统激发态的精确VMC方法的开发开辟了许多可能性,并大大扩展了神经网络波函数的应用范围。尽管只考虑了分子体系的电子激发和神经网络方法,但NES-VMC适用于任何量子哈密顿和任何方法,能够进行精确的计算研究,从而提高对振动耦合、光学带隙、核物理和其他具有挑战性的问题的理解。
总结
激发态在物理和化学的许多领域都很重要;然而,根据第一性原理对激发态性质进行可扩展、准确和鲁棒的计算仍然是一个关键的理论挑战。由深度学习驱动分子体系基态性质计算的最新进展表明,这种技术有可能解决这个问题。Pfau等人通过将变分量子蒙特卡罗直接推广到基态,提出了一种使用深度神经网络计算激发态的无参数数学原理。所提出的方法实现了对许多原子和分子的精确激发态计算,远远优于现有利用深度学习计算激发态性质的方法(特别是在较大的体系上),并且可以应用于各种量子体系。
—— Yury Suleymanov
2024年8月23日,Science在线发表了帝国理工学院David Pfau课题组的研究论文,题目为《Accurate computation of quantum excited states with neural networks》。
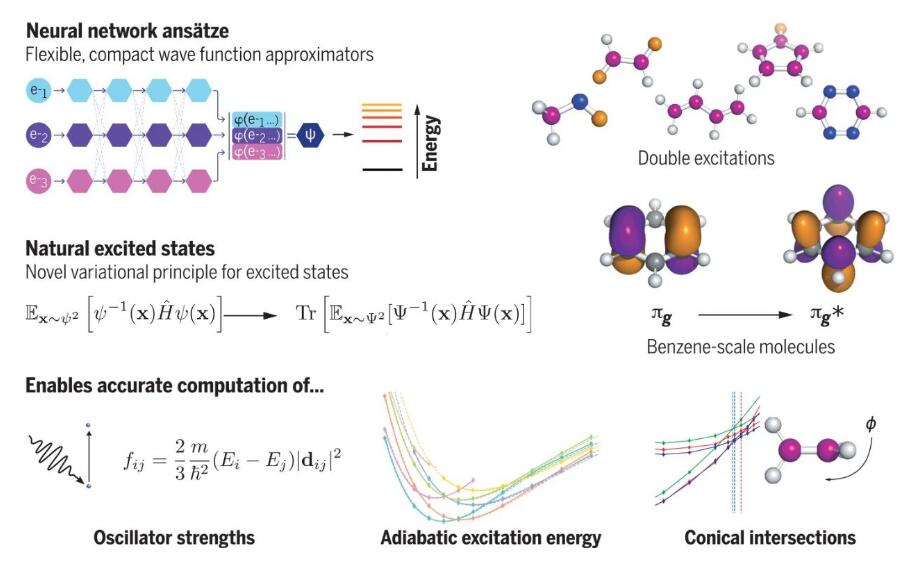
图1 自然激发态
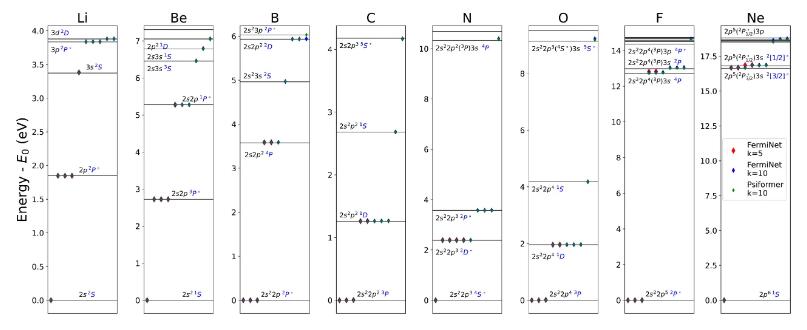
图2 从锂到氖的第一排原子的激发态能量

图3 小分子的垂直激发能和振子强度
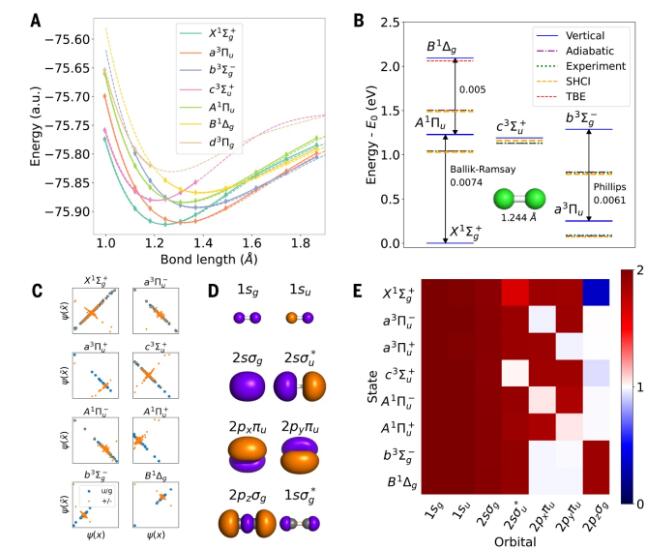
图4 碳二聚体的激发态
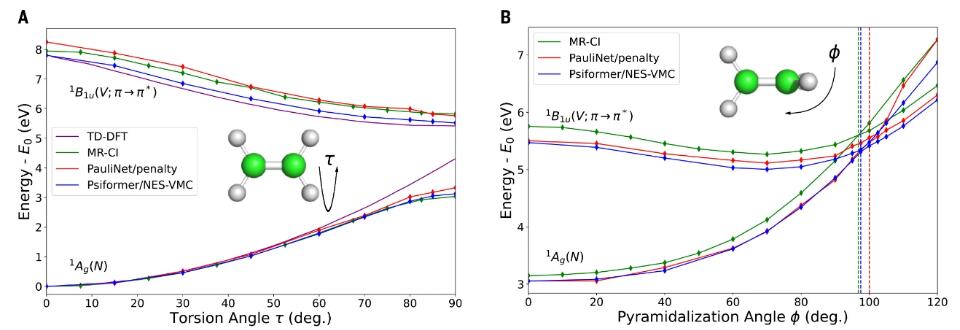
图5 乙烯的激发态和圆锥交点
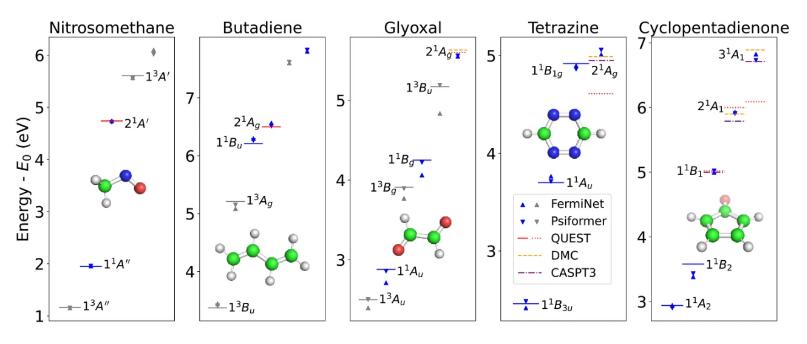
图6 大且双激发体系的激发态
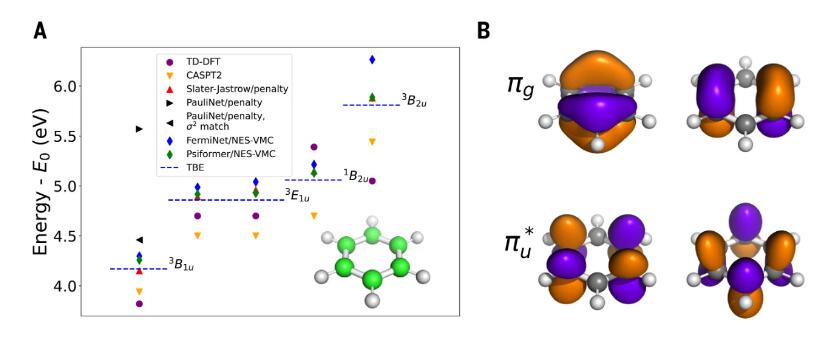
图7 苯的激发态
论文链接
Pfau, D., Axelrod, S., Sutterud, H. et al. Accurate computation of quantum excited states with neural networks. Science, 2024, 385, eadn0137. https://doi.org/10.1126/science.adn0137
其他相关文献
[1] Shaik, S., Danovich, D., Wu, W. et al. Quadruple bonding in C2 and analogous eight-valence electron species. Nat. Chem., 2012, 4, 195–200. https://doi.org/10.1038/nchem.1263
[2] Entwistle, M.T., Schätzle, Z., Erdman, P.A. et al. Electronic excited states in deep variational Monte Carlo. Nat. Commun., 2023, 14, 274. https://doi.org/10.1038/s41467-022-35534-5
来源:科研任我行 ,爱科会易仅用于学术交流