- 国内站
- 国际站
No data


仅收录五年以上有检索的国际会议
6月26日,《自然》(Nature)在线报道了武汉光电国家研究中心陈炜、刘宗豪团队题为「Buried interface molecular hybrid for inverted perovskite solar cells」的研究论文。

研究内容导读
反式(p-i-n)钙钛矿太阳能电池(perovskite solar cells, PSCs)因其兼顾高效率和稳定性、易于量产和叠层等优势,是当前PSCs这一新兴光伏技术产业化的主流技术路线。但在学术研究领域,正式(n-i-p)结构的PSCs的认证效率此前一直处于相对领先的位置,早期研究正式结构电池的学者更多。一直到2023年,得益于自组装单分子(Self‐assembled monolayers, SAMs)空穴选择层(hole selective layers, HSLs)和缺陷钝化策略的发展,反式PSCs的光电转换效率才超过正式PSCs。然而,常用的SAMs,如[4-(3,6-二甲基-9H-咔唑-9-基)丁基]膦酸(Me-4PACz),其本征导电性并不理想,器件效率对SAM分子的薄膜厚度极为敏感。这种分子在基底上的自组装状态不受控制、分子尺度上的分布不均会造成界面电荷传输损失,并且Me-4PACz对钙钛矿前驱体溶液的表面浸润性差,造成大量埋底界面微小孔洞和结晶不理想,从而导致大量埋底界面缺陷引发严重的界面复合,是限制反式PSCs效率进一步取得突破的重要原因。这些缺点,尤其是在制造大面积器件时将进一步放大。
针对上述问题,华中科技大学武汉光电国家研究中心陈炜-刘宗豪团队创新地提出一种埋底界面自组装单分子杂化(hybrid)策略,即在高性能自组装单分子Me-4PACz前驱液中引入同样具有大π共轭基团且含有对称多羧基的三苯胺单体(4,4’,4’’-硝基三苯甲酸(NA))。通过对比TA、BA等共吸附剂,发现NA作为共吸附剂,其分子结构更有利于增强与Me-4PACz间强π-π相互作用,能更好的减少Me-4PACz超薄膜在沉积过程中的自聚集效应,诱导Me-4PACz分子在2 nm尺度上获得更加均匀的分布,从而提高了钙钛矿薄膜埋底界面处光生载流子的抽取效率。
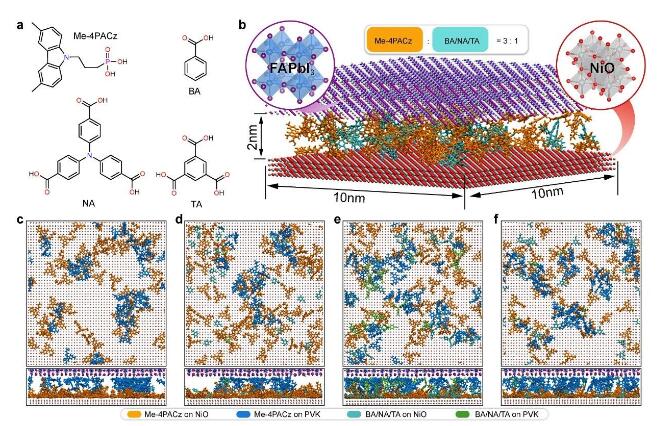
图1. 基于第一性原理计算的不同hybrid HSLs界面自组装状态NA诱导Me-4PACz实现最佳自组装成膜
另外,分子动力学模拟结果显示,Me-4PACz平趴式分布在氧化镍/钙钛矿界面,其膦酸基团和π环均可以与氧化镍基底作用,且Me-4PACz的π环能够钝化Vpb2+深能级陷阱,从而减少界面非辐射复合。不仅如此,多羧基NA单体的存在使得钙钛矿溶液在Me-4PACz上的润湿性得到了有效改善,消除了埋底界面处的纳米孔隙并释放了钙钛矿薄膜压缩应力,增强了钙钛矿埋底界面结晶性,这进一步降低了埋底界面缺陷浓度,且有利于大面积钙钛矿薄膜的均匀制备。
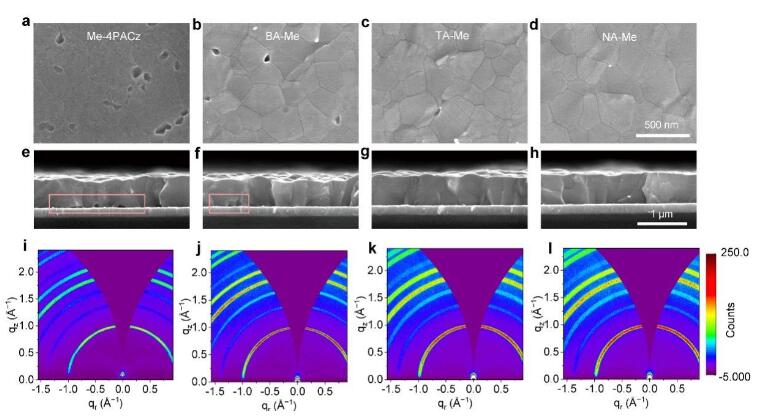
图2. 不同hybrid HSLs埋底界面处钙钛矿微孔形貌和结晶质量差异
综上,自组装单分子杂化空穴传输材料具有超浸润、纳米尺度均匀分布、载流子抽取速度快和非辐射复合低等优点,能够同时实现埋底界面载流子高效输运和缺陷钝化,大幅提升器件性能。基于择优带隙FA0.95Cs0.05PbI3 钙钛矿的反式PSCs在权威第三方机构(国家光伏产业计量测试中心NPVM)的准稳态认证效率高达26.54%,超过此前同机构认证的进表效率纪录(26.1%,NPVM)。
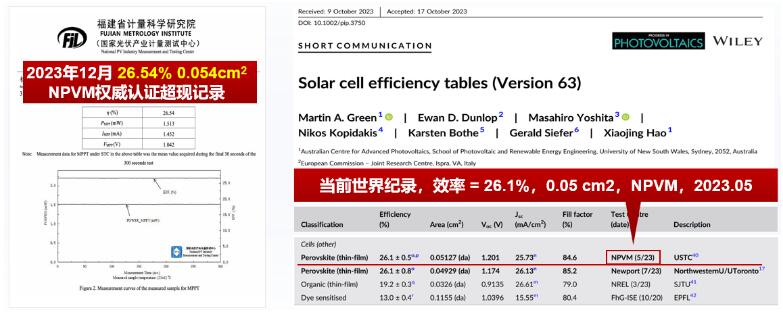
图3. 器件的权威认证效率超同期效率纪录
此外,该新型杂化自组装单分子材料良好的浸润性十分有利于制备大面积器件,在孔径面积为11.1 cm²的微型模组中实现了22.74%的认证效率,为反式微型模组国际同期最高认证效率,证明了埋底界面自组装分子杂化策略的可拓展性及其在大面积钙钛矿组件中的巨大应用前景。共吸附的多羧基单体增强了埋底界面处钙钛矿与NiO的键合连接,除效率提升外,也同步改善了器件的稳定性。目前上述研究成果已申请中国发明专利。
论文作者
华中科技大学是该论文的第一完成单位,华中科技大学刘三万博士、陈锐博士、孙振兴硕士、深圳职业技术大学霍夫曼先进材料研究院李竞白副教授、武汉理工大学肖文珊博士、和南方科技大学章勇助理教授为共同第一作者。华中科技大学陈炜教授、刘宗豪副教授和韩国成均馆大学Nam-Gyu Park教授为共同通讯作者。该研究工作得到了科技部国家重点研发计划项目、国家自然科学基金、华中科技大学自主创新研究基金、湖北省自然科学基金和光谷实验室创新计划等项目资助。
论文链接:
https://www.nature.com/articles/s41586-024-07723-3
6月26日,《自然》(Nature)在线报道了武汉光电国家研究中心陈炜、刘宗豪团队题为「Buried interface molecular hybrid for inverted perovskite solar cells」的研究论文。
研究内容导读
反式(p-i-n)钙钛矿太阳能电池(perovskite solar cells, PSCs)因其兼顾高效率和稳定性、易于量产和叠层等优势,是当前PSCs这一新兴光伏技术产业化的主流技术路线。但在学术研究领域,正式(n-i-p)结构的PSCs的认证效率此前一直处于相对领先的位置,早期研究正式结构电池的学者更多。一直到2023年,得益于自组装单分子(Self‐assembled monolayers, SAMs)空穴选择层(hole selective layers, HSLs)和缺陷钝化策略的发展,反式PSCs的光电转换效率才超过正式PSCs。然而,常用的SAMs,如[4-(3,6-二甲基-9H-咔唑-9-基)丁基]膦酸(Me-4PACz),其本征导电性并不理想,器件效率对SAM分子的薄膜厚度极为敏感。这种分子在基底上的自组装状态不受控制、分子尺度上的分布不均会造成界面电荷传输损失,并且Me-4PACz对钙钛矿前驱体溶液的表面浸润性差,造成大量埋底界面微小孔洞和结晶不理想,从而导致大量埋底界面缺陷引发严重的界面复合,是限制反式PSCs效率进一步取得突破的重要原因。这些缺点,尤其是在制造大面积器件时将进一步放大。
针对上述问题,华中科技大学武汉光电国家研究中心陈炜-刘宗豪团队创新地提出一种埋底界面自组装单分子杂化(hybrid)策略,即在高性能自组装单分子Me-4PACz前驱液中引入同样具有大π共轭基团且含有对称多羧基的三苯胺单体(4,4’,4’’-硝基三苯甲酸(NA))。通过对比TA、BA等共吸附剂,发现NA作为共吸附剂,其分子结构更有利于增强与Me-4PACz间强π-π相互作用,能更好的减少Me-4PACz超薄膜在沉积过程中的自聚集效应,诱导Me-4PACz分子在2 nm尺度上获得更加均匀的分布,从而提高了钙钛矿薄膜埋底界面处光生载流子的抽取效率。
图1. 基于第一性原理计算的不同hybrid HSLs界面自组装状态NA诱导Me-4PACz实现最佳自组装成膜
另外,分子动力学模拟结果显示,Me-4PACz平趴式分布在氧化镍/钙钛矿界面,其膦酸基团和π环均可以与氧化镍基底作用,且Me-4PACz的π环能够钝化Vpb2+深能级陷阱,从而减少界面非辐射复合。不仅如此,多羧基NA单体的存在使得钙钛矿溶液在Me-4PACz上的润湿性得到了有效改善,消除了埋底界面处的纳米孔隙并释放了钙钛矿薄膜压缩应力,增强了钙钛矿埋底界面结晶性,这进一步降低了埋底界面缺陷浓度,且有利于大面积钙钛矿薄膜的均匀制备。
图2. 不同hybrid HSLs埋底界面处钙钛矿微孔形貌和结晶质量差异
综上,自组装单分子杂化空穴传输材料具有超浸润、纳米尺度均匀分布、载流子抽取速度快和非辐射复合低等优点,能够同时实现埋底界面载流子高效输运和缺陷钝化,大幅提升器件性能。基于择优带隙FA0.95Cs0.05PbI3 钙钛矿的反式PSCs在权威第三方机构(国家光伏产业计量测试中心NPVM)的准稳态认证效率高达26.54%,超过此前同机构认证的进表效率纪录(26.1%,NPVM)。
图3. 器件的权威认证效率超同期效率纪录
此外,该新型杂化自组装单分子材料良好的浸润性十分有利于制备大面积器件,在孔径面积为11.1 cm²的微型模组中实现了22.74%的认证效率,为反式微型模组国际同期最高认证效率,证明了埋底界面自组装分子杂化策略的可拓展性及其在大面积钙钛矿组件中的巨大应用前景。共吸附的多羧基单体增强了埋底界面处钙钛矿与NiO的键合连接,除效率提升外,也同步改善了器件的稳定性。目前上述研究成果已申请中国发明专利。
论文作者
华中科技大学是该论文的第一完成单位,华中科技大学刘三万博士、陈锐博士、孙振兴硕士、深圳职业技术大学霍夫曼先进材料研究院李竞白副教授、武汉理工大学肖文珊博士、和南方科技大学章勇助理教授为共同第一作者。华中科技大学陈炜教授、刘宗豪副教授和韩国成均馆大学Nam-Gyu Park教授为共同通讯作者。该研究工作得到了科技部国家重点研发计划项目、国家自然科学基金、华中科技大学自主创新研究基金、湖北省自然科学基金和光谷实验室创新计划等项目资助。
论文链接:
https://www.nature.com/articles/s41586-024-07723-3


